
Metodi in silico per la valutazione della sicurezza dei cosmetici
Metodi in silico per la valutazione della sicurezza dei cosmetici
Un aiuto dal mondo dell’informatica
Alberto Manganaro • Kode Chemoinformatics, Pisa – a.manganaro@kode.srl
Di che cosa si parla
Con i metodi in silico si indicano un insieme di tecniche che permettono di ottenere informazioni su proprietà chimico-fisiche e attività biologiche di composti chimici, unicamente utilizzando strumenti informatici (il termine deriva dal parallelo con i test in vivo e in vitro). In particolare, uno dei metodi più rilevanti in tale ambito è rappresentato dalla Relazione quantitativa struttura-attività (Quantitative Structure-Activity Relationship, QSAR).
Il campo del QSAR, nato e usato nel mondo farmacologico per il drug design già da lungo tempo, si è notevolmente sviluppato negli ultimi decenni, trovando nuovi ambiti di applicazione. Nello specifico, negli ultimi anni è cresciuto un particolare interesse verso i QSAR per l’utilizzo ai fini delle valutazioni di proprietà (eco)tossicologiche, in particolare delle sostanze chimiche più in generale e degli ingredienti impiegati nelle formulazioni cosmetiche, nello specifico. Una spinta allo studio e all’approfondimento dei metodi in silico è sicuramente venuta anche dal fronte regolatorio: da una parte dal Regolamento (CE) n.1907/2006 (il Regolamento REACH), che motiva in modo conciso un principio essenziale dell’articolato, ovvero la volontà di promuovere lo sviluppo di metodi alternativi per la valutazione dei pericoli che le sostanze comportano; dall’altra perché viene ribadito che l’attuazione del Regolamento REACH dovrebbe basarsi sul ricorso, ogni volta che sia possibile, a metodi di prova alternativi atti a valutare i pericoli che le sostanze chimiche comportano per la salute e per l’ambiente. Questo fa sì che l’uso degli animali (ovvero dell’esecuzione di test in vivo) dovrebbe essere evitato ricorrendo a metodi alternativi validati dalla Commissione, da organismi internazionali oppure riconosciuti dalla Commissione o dall’Agenzia come idonei a soddisfare le prescrizioni in materia di informazione imposte da REACH. Anche il Regolamento (CE) n.1272/2008 (CLP) rincalza su questo fronte, affermando che qualora debbano essere effettuati nuovi test sperimentali al fine di indagare una proprietà intrinseca di una sostanza/miscela (per poterne stabilire la classificazione e la conseguente etichettatura), le prove sugli animali vengono effettuate soltanto se non esistono alternative che offrano adeguata attendibilità e qualità dei dati. A questi si affianca, considerando il mero ordine cronologico dei regolamenti europei, il Regolamento (CE) n.1223/2009 (sui prodotti cosmetici), dove è stato fissato un calendario delle scadenze, in corrispondenza delle quali viene fatto divieto di commercializzare prodotti cosmetici la cui formulazione finale, i cui ingredienti o combinazioni di ingredienti siano stati testati su animali.
In questo scenario emerge la rilevanza degli strumenti in silico, per poter ottenere dati spesso fondamentali per la commercializzazione dei prodotti chimici e cosmetici, evitando i test su animali. Le stesse aziende (in particolare medie e piccole) guardano con molto interesse ai QSAR, dato che l’uso di strumenti predittivi in silico risulta più veloce e soprattutto più economico del test diretto. Tale interesse viene anche espresso a livello europeo, dove sono sempre maggiori i progetti finalizzati allo sviluppo dei QSAR e al loro concreto utilizzo in ambito normativo. Il Regolamento REACH, come menzionato prima, ad esempio parla proprio di una strategia comunitaria di promozione di metodi di prova alternativi, considerandola una priorità: a questo proposito, la Commissione, gli Stati membri, l’industria e gli altri soggetti interessati dovrebbero continuare a contribuire alla promozione, a livello internazionale e nazionale, di metodi di prova alternativi, tra cui metodologie assistite dal computer.
Ma per poter utilizzare tali strumenti e rendere i loro risultati accettabili a fini normativi, è necessario che questi vengano maneggiati con cognizione di causa dato che, come tutti gli strumenti, hanno vantaggi e potenzialità, ma anche limiti che vanno compresi.
Come lavorano i QSAR?
Nel QSAR si sviluppano modelli matematici che legano la struttura teorica di un composto a una determinata proprietà di interesse. Questo viene fatto attraverso diverse tecniche di Machine Learning che per costruire il modello hanno bisogno di un insieme di dati noti per l’apprendimento. Dunque, in parole semplici, il punto di partenza è un insieme di molecole per le quali è noto sperimentalmente il valore della proprietà di interesse e questo insieme viene usato per apprendere quali caratteristiche delle strutture chimiche possono essere legate a valori maggiori o minori della proprietà (e infatti l’insieme viene chiamato training set). Questo processo di apprendimento porta alla creazione dello specifico modello che sarà poi in grado di fornire una predizione della proprietà per qualsiasi nuova struttura sia fornita in input.
Quindi i QSAR sono modelli matematici costruiti attraverso un processo di estrazione di informazioni utili da un insieme di dati noti di apprendimento. Sono modelli predittivi basati su informazioni estratte statisticamente, per cui il modo in cui specifici aspetti delle strutture molecolari di origine sono legati alla proprietà di interesse non è necessariamente interpretabile da un punto di vista chimico o tossicologico. Nonostante questo, quando il modello è costruito utilizzando tecniche robuste e soprattutto specifici metodi di validazione, le predizioni fornite rappresentano un’informazione affidabile anche quando non direttamente interpretabile.
Questo non toglie che in molti QSAR sia inserita anche la conoscenza pregressa di carattere chimico e tossicologico.
In questi casi le caratteristiche delle molecole da prendere in considerazione sono note a priori e sono la base del modello (da sole o unite con altre caratteristiche estratte con metodi di Machine Learning). Ad esempio, alcuni modelli noti per predire l’attività di mutagenesi di un composto (nello specifico, predire il risultato del test di Ames) si basano su una trentina di regole chiamate Structural Alerts, note e studiate in letteratura scientifica, e che pertanto non solo sono ben definite (tipicamente sono descritte dalla presenza o meno di un particolare gruppo funzionale, ad esempio gruppi nitro-aromatici) ma hanno anche una spiegazione del meccanismo di azione (e quindi è documentato il modo in cui la presenza di un determinato gruppo funzionale rende il composto mutageno) (Fig.1).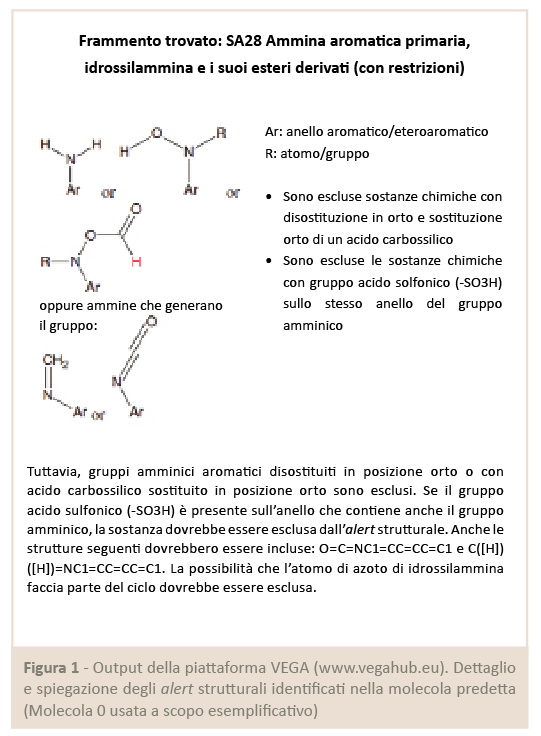
Dall’altro lato, le caratteristiche strutturali estratte e usate dagli algoritmi di Machine Learning (quindi in modo puramente statistico) possono ricevere, a posteriori, una spiegazione meccanicistica e fornire un utile input agli esperti del dominio che si occupano di chimica e tossicologia.
In tal modo si va a formare un “circolo virtuoso”, per cui le informazioni derivanti da un approccio statistico alimentano studi scientifici sui possibili modi di azione di tossicità dei composti e tale conoscenza a sua volta rientrerà nei successivi modelli QSAR sviluppati.
La validazione dei modelli in silico
Si accennava ai metodi di validazione dei modelli: tali informazioni risultano molto importanti per la valutazione sulle possibilità di utilizzo dei QSAR, in quanto forniscono a priori un’indicazione di quanto possano essere accurati. I modelli vengono tipicamente resi pubblici e corredati da statistiche riguardo le capacità predittive; proprio per avere una reale valutazione di quanto un modello possa essere predittivo, andrebbero sempre osservati con attenzione i valori relativi alla sua validazione. Di solito sono forniti prima di tutto valori riferiti al training set del modello stesso; tali valori possono essere ad esempio per modelli che forniscono una classificazione: quelli dell’accuratezza (numero totale di predizioni corrette sul totale delle predizioni fatte), della sensibilità (numero di predizioni positive corrette sul totale dei composti positivi/tossici) e della specificità (numero di predizioni negative corrette sul totale dei composti negativi/non tossici). Ma tali valori, riferiti allo stesso training set sul quale è stato costruito il modello, possono essere ingannevoli; un modello può avere delle performance molto buone sugli stessi dati su cui è stato costruito, ma risultare scarsamente affidabile su qualsiasi altro nuovo composto che si voglia predire. Questa situazione viene definita di overfitting e si ha quando il modello è così tanto “aderente” ai dati su cui è costruito da non essere in grado di essere generalizzabile (e quindi utilizzabile) a nuovi composti.
Per questo uno degli approcci più comuni di validazione è l’utilizzo di un cosiddetto test set: un insieme di molecole per le quali è noto ovviamente il valore sperimentale della proprietà e che non viene utilizzato in alcun modo nella costruzione del modello, ma al quale si applica a posteriori il modello sviluppato proprio per analizzare le statistiche che avrà su un insieme di dati esterno. Per cui, il valore di accuratezza sul test set risulterà molto utile perché rappresenta un’indicazione di quali performance potrebbe avere il modello nel suo utilizzo reale su dei nuovi composti.
Il dominio di applicabilità di un modello
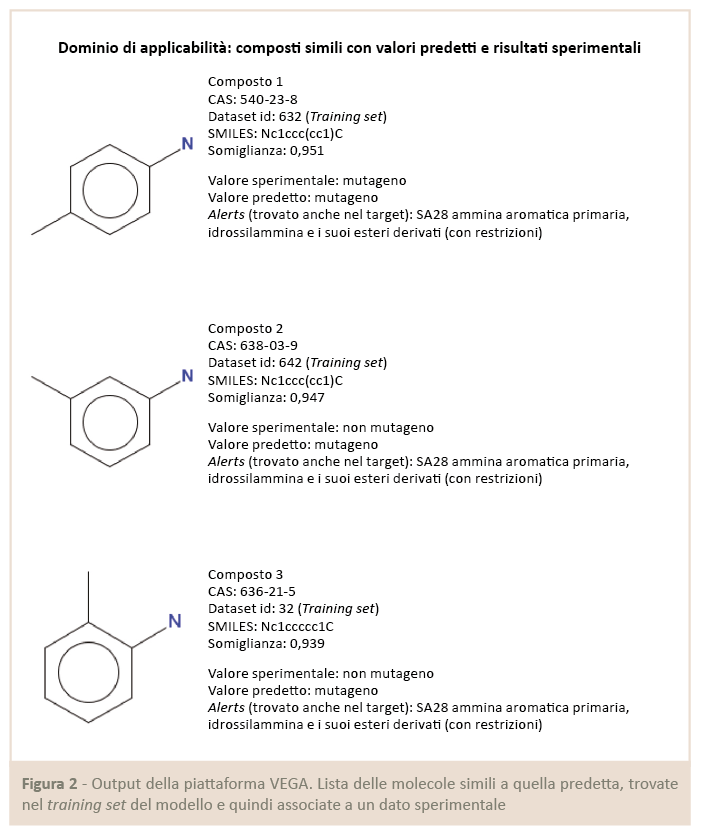 Le indicazioni fornite fino a ora riguardano le performance generiche di un QSAR, ma per la valutazione dell’uso concreto di un modello vanno affiancate al concetto di dominio di applicabilità. Esso serve a fornire un’informazione specifica di quanto possa essere affidabile la singola predizione fatta su un determinato composto.
Le indicazioni fornite fino a ora riguardano le performance generiche di un QSAR, ma per la valutazione dell’uso concreto di un modello vanno affiancate al concetto di dominio di applicabilità. Esso serve a fornire un’informazione specifica di quanto possa essere affidabile la singola predizione fatta su un determinato composto.
Il dominio di applicabilità consiste in una definizione dello spazio chimico su cui si è costruito il modello e quindi descrive il tipo di composti che costituiscono il training set (Fig.2).
Se un nuovo composto non rientra in questo spazio, ovvero ha caratteristiche chimico-strutturali particolarmente diverse dai composti del training set, il modello non potrà fornire una predizione affidabile su di esso, perché starà fondamentalmente fornendo un’estrapolazione. Ad esempio, per un modello costruito con un training set dove nessun composto è alogenato, un nuovo composto da predire contenente degli alogeni risulterebbe fuori dal dominio di applicabilità; quindi la sua predizione risulterebbe poco affidabile e si può dire che il modello non sia adatto a lavorare su composti di quella famiglia chimica.
Attenzione ai risultati!
Da quanto detto fin qui risulta chiaro che i QSAR possono essere uno strumento molto potente, ma non vanno intesi come una black-box che fornisce valori da prendere così come vengono prodotti. Ciascuna predizione va valutata sia sulla base delle caratteristiche a priori del modello sia su indicatori che riguardano la specifica predizione per il composto fornito in input. Quindi i modelli QSAR non sostituiscono la valutazione dell’esperto di dominio; al contrario, forniscono proprio all’esperto una serie di informazioni importanti che contribuiscono alla sua valutazione finale. Non a caso si utilizza spesso il termine Weight of Evidence (WoE) per indicare un approccio in cui una valutazione di tossicità o di altra attività viene fatta senza ricorrere a test in vivo ad hoc, ma sulla base di una serie di evidenze quali l’utilizzo di diversi modelli QSAR (costruiti su training set diversi e con metodi differenti) unito a ricerche di letteratura, nonché a ricerche di composti noti analoghi del composto di interesse, in modo che l’esperto possa fornire con una certa sicurezza una valutazione finale basata su una molteplicità di dati e informazioni.
Per questo un buono strumento QSAR fornisce all’utente più informazioni e indicazioni possibili, oltre il mero valore predetto (Fig.3).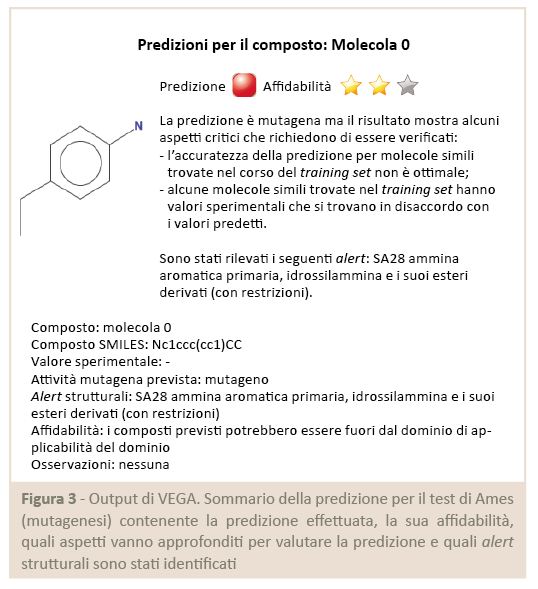
Si noti che i margini di incertezza non rendono necessariamente questi strumenti meno validi dei test in vivo. Infatti va ricordato che anche i risultati della sperimentazione diretta sono soggetti a variabilità derivante da molteplici fattori, a partire dalla semplice variabilità sperimentale (che nel caso di test tossicologici su pesci o mammiferi può anche essere rimarcabilmente alta). Per tale motivo, l’esortazione a non usare ciecamente i risultati è valida per ogni tipo di valore, indipendentemente dalla sua fonte (in silico, in vivo o in vitro).
A termine della rapida panoramica sul QSAR può essere interessante accennare, a titolo di esempio concreto degli strumenti disponibili, alla piattaforma VEGA (www.vegahub.eu), strumento gratuito sviluppato da Kode Chemoinformatics e dall’Istituto di Ricerche Farmacologiche “Mario Negri” all’interno di svariati progetti di ricerca. Questo strumento risulta di utilizzo molto semplice: l’input necessario è la struttura molecolare fornita in uno dei possibili formati digitali (come stringa SMILES o file SDF) e la selezione delle proprietà di interesse fra quelle disponibili (la lista è in continuo ampliamento dato che nuovi modelli vengono continuamente sviluppati e resi disponibili). Il risultato prodotto è un report completo nel quale, proprio alla luce di quanto spiegato in precedenza, non viene riportato il semplice valore della predizione ma una serie di informazioni accessorie grazie alle quali è possibile valutare appieno l’affidabilità del risultato.
Seguendo questo stesso approccio e sfruttando l’architettura di VEGA, si stanno sviluppando anche strumenti per alcuni ambiti specifici. Ad esempio, all’interno del progetto europeo LIFE VERMEER (LIFE16 ENV/IT/000167) è in lavorazione (e disponibile, sempre gratuitamente, a breve) un’applicazione per produrre valutazioni utili ai fini normativi specifiche per gli ingredienti cosmetici. Per approfondimenti su di essa si rimanda a una prossima pubblicazione seguente al rilascio ufficiale del software (entro la fine del 2020).
